Примерная продолжительность: 60 минут
Цели обучения
- Изучить различные типы методов нормализации
- Познакомиться с объектами данных
DESeqDataSet - Понять, как нормализовать каунты с помощью DESeq2
Нормализация
Первым шагом в процедуре DE-анализа является нормализация числа прочтений, которая необходима для точного сравнения экспрессии генов между образцами.
Количество картированных на геном прочтений для каждого гена (каунтов) пропорционально экспрессии РНК (“интересные”) в дополнение ко многим другим факторам (“неинтересные”). Нормализация - это процесс масштабирования необработанных значений подсчета для того, чтобы учесть “неинтересные” факторы. Таким образом, уровни экспрессии становятся более сопоставимыми между образцами и/или внутри образцов.
Основными факторами, которые часто учитываются при нормализации, являются:
-
Глубина секвенирования: Учет глубины секвенирования необходим для сравнения экспрессии генов между образцами. В приведенном ниже примере экспрессия каждого гена в образце А по сравнению с образцом В увеличилась в два раза, однако это является следствием того, что в образце А* глубина секвенирования в два раза больше.
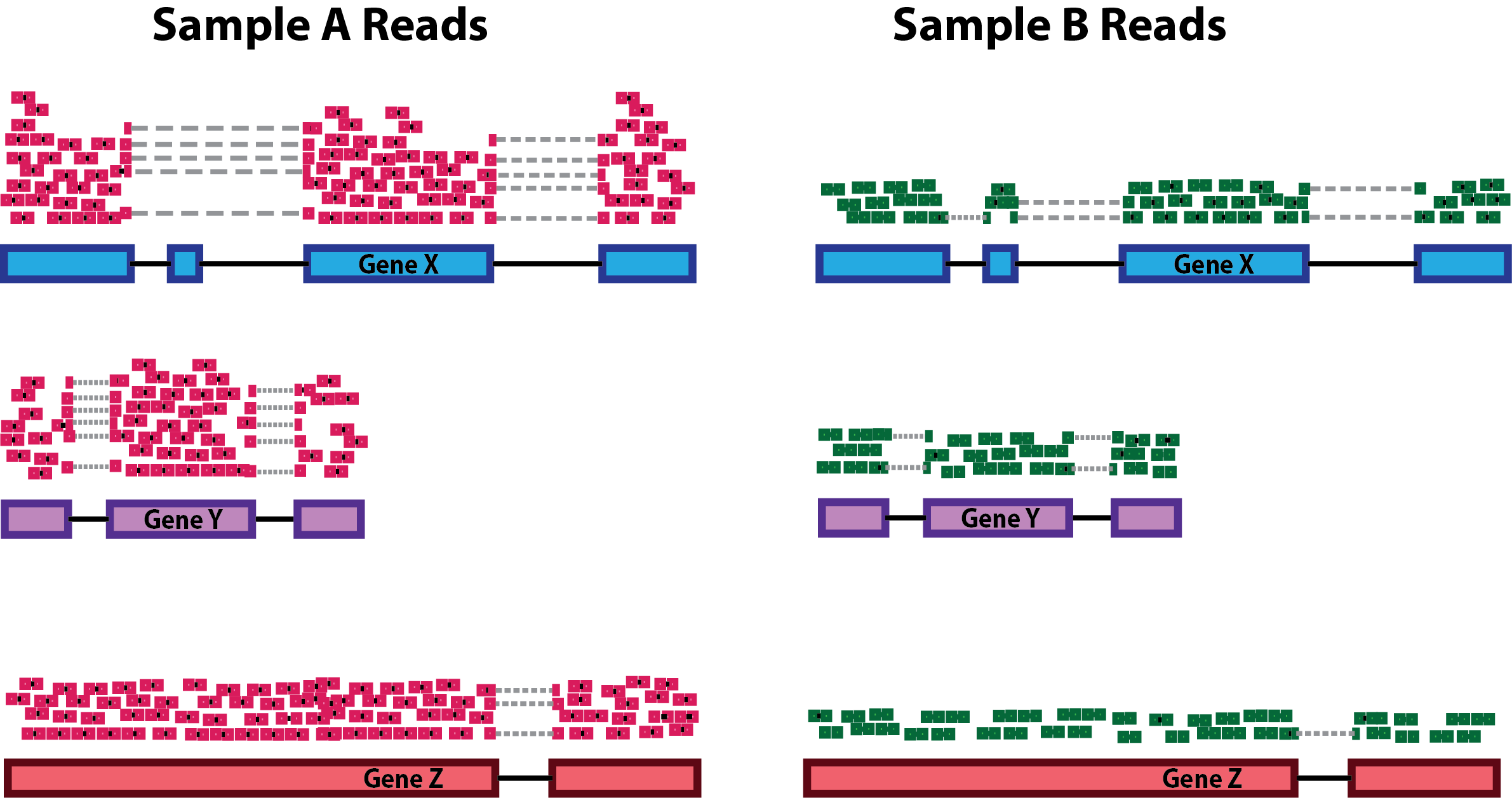
NOTE: На рисунке выше каждый розовый и зеленый прямоугольник представляет собой рид, выровненное на ген. Прямоугольники, соединенные пунктирными линиями, обозначают риды, перекрывающие стыки между эксзонами.
-
Длина гена: Учитывать длину гена необходимо для сравнения экспрессии разных генов в одном и том же образце. В примере ген X и ген Y имеют схожие уровни экспрессии, но количество считываний, сопоставленных с геном X, будет намного больше, чем с геном Y, поскольку ген X длиннее.
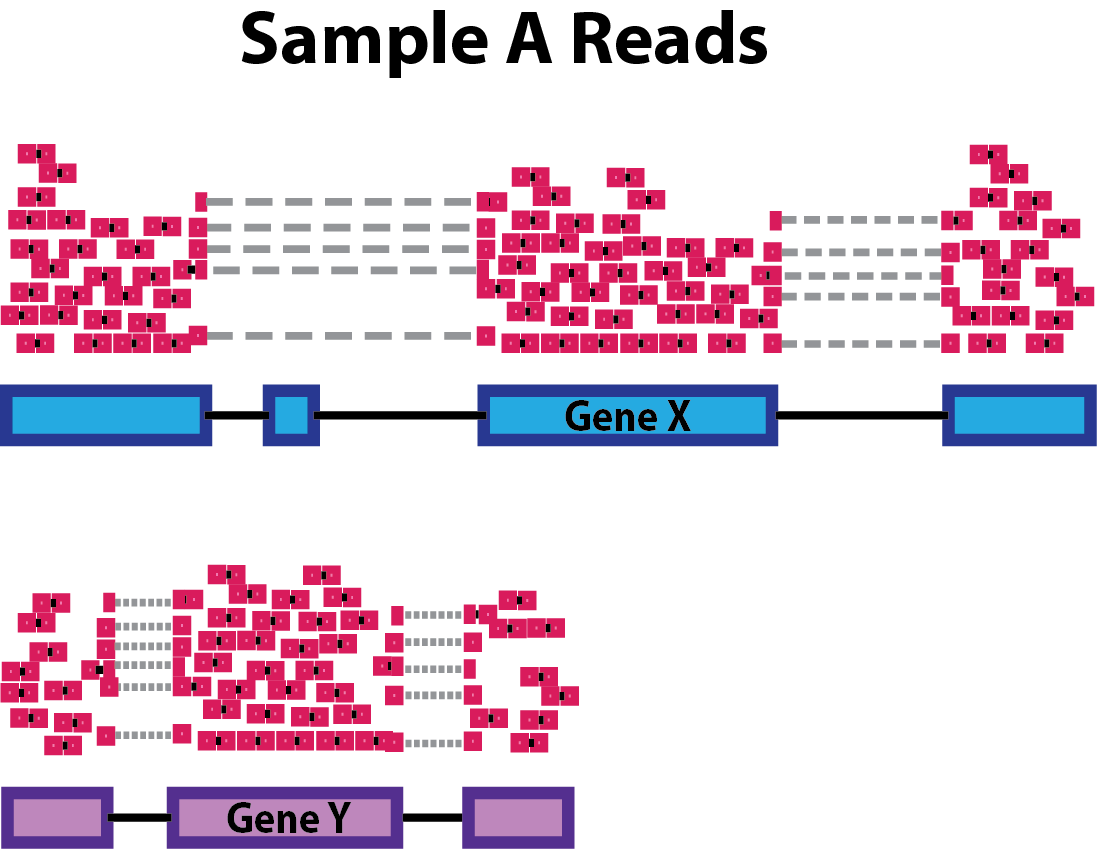
-
Состав РНК: Небольшое количество генов с сильной дифференциальная экспрессей между образцами, различия в количестве экспрессируемых генов между образцами или присутствие контаминации могут привести к искажению некоторых методов нормализации. Для точного сравнения экспрессии между образцами рекомендуется учитывать состав РНК, что особенно важно при проведении анализа дифференциальной экспрессии [1].
В примере, если бы мы разделили каждый образец на общее количество прочтений для нормализации, число прочтений было бы сильно искажено из-за DE-гена, на который приходится большая часть прочтений для образца A, но не для образца B. Большинство других генов для образца А будут разделены на более высокое число суммарных прочтений и окажутся менее экспрессированными, чем те же гены в образце В.

Хотя нормализация важна для анализа дифференциальной экспрессии, она также необходима для разведочного анализа, визуализации данных и в других случаях, когда вы исследуете или сравниваете прочтения между или внутри образцами.
Общие методы нормализации
Для учета указанных различий существует несколько распространенных методов нормализации:
| Метод нормализации | Описание | Учитываемый фактор | Рекоммендации по использованию |
|---|---|---|---|
| CPM (число прочтений на миллион) | число прочтений для гена поделенное на общее число ридов | глубина секвенирования | сравнение числа прочтений для гена между повторами из одной и той же группы образцов; НЕ для сравнения внутри образца или DE-анализа |
| TPM (Число транскриптов на килобазу на миллион) | Число прочтений, деленое на длинну транскрипта в килобазах на миллион прокартированных ридов | глубина секвенирования и число ридов | Сравнение числа прочтений для генов внутри образца или между образцами из одной группы; Не для DE-анализа |
| RPKM/FPKM (риды/фрагменты (риды, секвенированные с двух сторон) на килобазу экзона на миллион прокартированных ридов/фрагментов) | То же, что и TPM | глубина секвенирования и длина гена | сравнение количества прочтений между генами внутри образца; Не для сравнения между образцами или DE-анализа |
| Медиана отношений из пакета DESeq2’s [1] | Число прочтений, деленое на специфичный для каждого образца размерный фактор (size factor), который определяется как вычисланная по всем генам в образце медиана отношения среднего числа прочтений гена в образце к среднему геометрическому числу прочтений этого гена между образцами | глубина секвенирования и состав РНК | сравнение числа прочтений гена между образцами и DE-анализ; Не для сравнения генов внутри образца |
| Усеченное среднее М-значений (TMM) из пакета EdgeR [2] | использует взвешенные усеченные средние логарифма отношения экспрессии между образцами | глубина секвенирования, состав РНК и длина гена | сравнение числа прочтений внутри и между образцами, а также DE-анализ |
RPKM/FPKM (не рекомендуется)
Хотя методы нормализации TPM и RPKM/FPKM учитывают глубину секвенирования и длину гена, RPKM/FPKM не рекомендуется использовать. Причина в том, что нормализованные значения прочтений, полученные методом RPKM/FPKM, несопоставимы между образцами.
При использовании нормализации RPKM/FPKM общее количество нормализованных прочтений RPKM/FPKM для каждого образца будет разным. Поэтому вы не можете в равной степени сравнивать нормализованные прочтения для каждого гена в между образцами.
RPKM-нормализованная таблица прочтений
| ген | образецA | образецB |
|---|---|---|
| XCR1 | 5.5 | 5.5 |
| WASHC1 | 73.4 | 21.8 |
| … | … | … |
| Общая количество RPKM-нормализованных прочтений | 1,000,000 | 1,500,000 |
Например, в приведенной выше таблице в образцеА имеется более высокая доля прочтений, связанных с XCR1 (5,5/1 000 000), чем в образцеВ (5,5/1 500 000), несмотря на то, что величины прочтений RPKM одинаковы. Поэтому мы не можем напрямую сравнить количество прочтений для XCR1 (или любого другого гена) между образцами А и В, так как общее количество нормализованных прочтений в разных образцах разное.
Нормализованные по DESeq2 прочтения: метод медианы отношения
Поскольку инструменты для дифференциального анализа экспрессии сравнивают каунты между группами образцов для одного и того же гена, длина гена не должна учитываться инструментом. Однако глубина секвенирования и состав РНК должны быть приняты во внимание.
Для нормализации глубины секвенирования и состава РНК в DESeq2 используется метод медианы отношений. С точки зрения пользователя это только одна команда, но на програмном уровне это несколько шагов, описанных ниже.
NOTE: Ниже подробно описаны некоторые шаги, выполняемые DESeq2 при запуске одной функции для получения DE-генов. В принципе, для типичного анализа РНК-секвенирования вы не будете выполнять эти шаги по отдельности..
Шаг 1: создание псевдореференcной выборки (среднее геометрическое значение строк)
Для каждого гена (то есть каджой строки) создается псевдореференсный (pseudo-reference) образец, равный среднему геометрическому по всем образцам.
| ген | образецА | образецB | псевдорефенсный образец |
|---|---|---|---|
| EF2A | 1489 | 906 | sqrt(1489 * 906) = 1161.5 |
| ABCD1 | 22 | 13 | sqrt(22 * 13) = 17.7 |
| … | … | … | … |
Шаг 2: вычисление отношения каждого образца к референсу
Для каждого гена в образце рассчитываются соотношения (образец/референс) (как показано ниже). Это выполняется для каждого образца в наборе данных. Поскольку большинство генов не являются дифференциально экспрессируемыми, большинство генов в каждом образце должны иметь схожие соотношения внутри образца.
| ген | образецА | образецB | псевдореференсный образец | отношение образецA/псевдореференс | отношение образецВ/псевдореференс |
|---|---|---|---|---|---|
| EF2A | 1489 | 906 | 1161.5 | 1489/1161.5 = 1.28 | 906/1161.5 = 0.78 |
| ABCD1 | 22 | 13 | 16.9 | 22/16.9 = 1.30 | 13/16.9 = 0.77 |
| MEFV | 793 | 410 | 570.2 | 793/570.2 = 1.39 | 410/570.2 = 0.72 |
| BAG1 | 76 | 42 | 56.5 | 76/56.5 = 1.35 | 42/56.5 = 0.74 |
| MOV10 | 521 | 1196 | 883.7 | 521/883.7 = 0.590 | 1196/883.7 = 1.35 |
| … | … | … | … |
Шаг 3: вычисление коэффициента нормализации для каждого образца ( размерный фактор).
Медианное значение всех отношений для данного образца (медианное значение столбца в таблице выше) берется в качестве коэффициента нормализации (размерного фактора) для этого образца, как рассчитано ниже. Обратите внимание, что дифференциально экспрессированные гены не должны влиять на медианное значение:
normalization_factor_sampleA <- median(c(1.28, 1.3, 1.39, 1.35, 0.59))
normalization_factor_sampleB <- median(c(0.78, 0.77, 0.72, 0.74, 1.35))
На рисунке ниже показано медианное значение для распределения соотношений по всем генам внутри одного образца (частота находится на оси y).
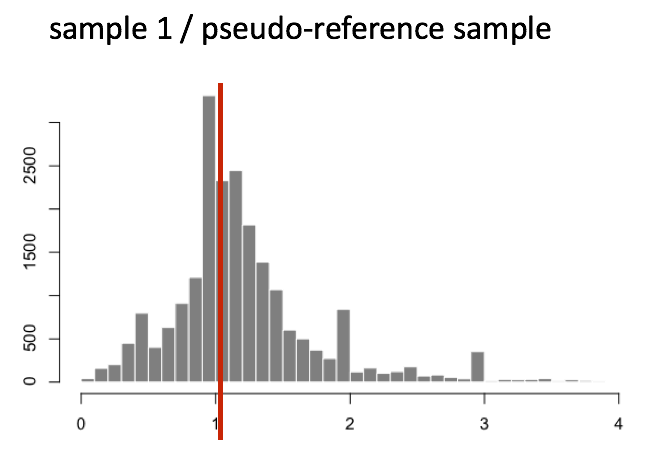
Метод медианы соотношений предполагает, что не ВСЕ гены дифференциально экспрессируются, поэтому коэффициенты нормализации должны учитывать глубину секвенирования и состав РНК образца (большие выпадающие гены не будут представлять медианные значения соотношений). Этот метод устойчив к дисбалансу в up-/down-регуляции и большому количеству дифференциально экспрессируемых генов.
Обычно эти размерные факторы находятся около 1, если вы видите большие различия между образцами, важно обратить на это внимание, так как это может указывать на наличие экстремальных выбросов.
Шаг 4: вычисление нормализованных каунтов с использованием коэффициента нормализации
Для получения нормализованных каунтов, каждое необработанное количество прочтений в данном образце делится на коэффициент нормализации для этого образца. Это выполняется для каждого гена во всех образцах. Например, если медианный фактор для образцаА равен 1,3, а медианный фактор для образцаВ равен 0,77, то нормализованные значения можно рассчитать следующим образом:
SampleA median ratio = 1.3
SampleB median ratio = 0.77
Необработанные (raw) каунты
| ген | образецA | образецB |
|---|---|---|
| EF2A | 1489 | 906 |
| ABCD1 | 22 | 13 |
| … | … | … |
Нормализованные каунты
| ген | образецA | образецB |
|---|---|---|
| EF2A | 1489 / 1.3 = 1145.39 | 906 / 0.77 = 1176.62 |
| ABCD1 | 22 / 1.3 = 16.92 | 13 / 0.77 = 16.88 |
| … | … | … |
Обратите внимание, что нормализованные каунты не являются целыми числами.
Упражнение
Определите нормализованные каунты для гена PD1, учитывая приведенные ниже необработанные каунты и размерные факторы.
ПРИМЕЧАНИЕ: Вам нужно будет выполнить приведенный ниже код для создания таблицы данных с необработанными каунтами (PD1) и вектора размерных факторов (size_factors), а затем использовать эти объекты для определения значений нормализованных каунтов:
# Необработанные каунты для PD1
PD1 <- c(21, 58, 17, 97, 83, 10)
names(PD1) <- paste0("Sample", 1:6)
PD1 <- data.frame(PD1)
PD1 <- t(PD1)
# Размерные факторы для каждого образца
size_factors <- c(1.32, 0.70, 1.04, 1.27, 1.11, 0.85)
Нормализация каунтов для датасета Mov10 с использованием DESeq2
Теперь, когда мы знаем теорию нормализации числа прочтений, мы нормализуем каунты для набора данных Mov10 с помощью DESeq2. Для этого необходимо выполнить несколько шагов:
- Убедитесь, что имена строк таблицы метаданных присутствуют и расположены в том же порядке, что и имена столбцов таблицы числа прочтений.
- Создайте объект
DESeqDataSet - Сгенерируйте нормализованные каунты
1. Соответствие метаданных и данных числа прочтений
Мы всегда должны следить за тем, чтобы имена образцов совпадали в двух файлах и чтобы образцы располагались в правильном порядке. Если это не так, DESeq2 выдаст ошибку.
### Проверяем, что названия образцов совпадают в обоих файлах
all(colnames(data) %in% rownames(meta))
all(colnames(data) == rownames(meta))
Если ваши данные не совпали, вы можете использовать функцию match(), чтобы переставить их так, чтобы они совпали.
Упражнение
Предположим, что в матрице числа прочтений и файле метаданных имена образцов совпадают, но они расположены не по порядку. Напишите строку(и) кода, необходимую для создания новой матрицы, столбцы которой упорядочены таким образом, чтобы они совпадали с именами строк метаданных.
2. Create DESEq2 object
Программные пакеты Bioconductor часто определяют и используют пользовательский класс в R для хранения данных (входных данных, промежуточных данных, а также результатов). Эти пользовательские структуры данных похожи на списки тем, что они могут содержать несколько различных типов/структур данных. Но, в отличие от списков, они имеют заранее определенные слоты данных, которые содержат конкретные типы/классы данных. Доступ к данным, хранящимся в этих заранее определенных слотах, можно получить с помощью специальных функций, определенных пакетом.
Давайте начнем с создания объекта DESeqDataSet, а затем поговорим немного подробнее о том, что хранится внутри него. Для создания объекта нам понадобятся матрица count и таблица metadata в качестве исходных данных. Нам также нужно будет указать формулу дизайна. Формула дизайна определяет столбец (столбцы) в таблице метаданных и то, как они должны использоваться в анализе. Для нашего набора данных у нас есть только один интересующий нас столбец, это ~sampletype. Этот столбец имеет три уровня факторов, что говорит DESeq2, что для каждого гена мы хотим оценить изменение экспрессии генов относительно этих разных уровней.
## Создаем объект DESeq2Dataset
dds <- DESeqDataSetFromMatrix(countData = data, colData = meta, design = ~ sampletype)

При желании вы можете использовать специльно созданные в DESeq функции для доступа к различным слотам и получения информации. Например, если бы мы хотели получить исходную матрицу числа прочтений, мы бы использовали функцию counts() (Примечание: мы вложили ее в функцию View(), так что вместо вывода в консоль мы можем увидеть ее в редакторе скриптов):
View(counts(dds))
По мере прохождения рабочего процесса мы будем использовать соответствующие функции для проверки того, какая информация хранится внутри нашего объекта.
3. Генерация нормализованных каунтов для the Mov10
Следующим шагом является нормализация данных о числе прочтений, чтобы можно было провести справедливое сравнение генов между образцами.
Для выполнения нормализации методом медианы отношений в DESeq2 есть единственная функция estimateSizeFactors(), которая генерирует для нас размерные факторы. Мы будем использовать эту функцию в приведенном ниже примере, но в типичном анализе RNA-seq этот шаг автоматически выполняется функцией DESeq(), которую мы рассмотрим позже.
dds <- estimateSizeFactors(dds)
Присваивая результаты обратно объекту dds, мы заполняем слоты объекта DESeqDataSet соответствующей информацией. Мы можем посмотреть на коэффициент нормализации, примененный к каждому образцу, используя:
sizeFactors(dds)
Теперь, чтобы получить нормализованную матрицу подсчетов из dds, мы используем функцию counts() и добавим аргумент normalized=TRUE.
normalized_counts <- counts(dds, normalized=TRUE)
Мы можем сохранить эту нормализованную матрицу данных в файл для последующего использования:
write.table(normalized_counts, file="data/normalized_counts.txt", sep="\t", quote=F, col.names=NA)
NOTE: DESeq2 на самом деле не использует нормализованные каунты, вместо этого он использует необработанные каунты и моделирует нормализацию внутри обобщенной линейной модели (GLM). Эти нормализованные подсчеты будут полезны для последующей визуализации результатов, но не могут быть использованы в качестве входных данных для DESeq2 или любых других инструментов, выполняющих анализ дифференциальной экспрессии, которые используют отрицательную биномиальную модель.
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.